写在前面
在上篇文章中,我们学习了PCA分析基本过程,接下来接着学习系统发育树的构建和祖先成分分析。
二、 邻接法构建系统发生树
基于SNP数据构建系统发生树,省略了序列比对的步骤,这里推荐使用邻接法(Neighbour-Joining,NJ)。
1. 简单介绍邻接法和系统发生树
系统发生树(Phylogenetic tree)又称为演化树(evolutionary tree),是表明被认为具有共同祖先的各物种间演化关系的树,是一种亲缘分支分类方法(cladogram)。在树中,每个节点代表其各分支的最近共同祖先,而节点间的线段长度对应演化距离(如估计的演化时间)
NJ法是基于最小进化原理经常被使用的一种算法,它不检验所有可能的拓扑结构,能同时给出拓扑结构和分支长度。在重建系统发育树时,它取消了UPGMA法所做的假定,认为在进化分支上,发生趋异的次数可以不同。该方法通过确定距离最近(或相邻)的成对分类单位来使系统树的总距离达到最小。它的特点是重建的树相对准确,假设少,计算速度快,只得到一棵树。
2. 数据处理和准备
- 过滤缺失率较高(–geno 0.05)和强连锁SNP位点(–indep-pairwise)
#!/bin/bash
plink=/software/biosoft/software/plink/plink
time $plink --vcf 60_dog.merge.vcf --geno 0.05 --dog --autosome --set-missing-var-ids @:# --indep-pairwise 50 10 0.3 --out 60_dog && echo "----prune SNP done----"
time $plink --vcf 60_dog.merge.vcf --geno 0.05 --dog --autosome --set-missing-var-ids @:# --extract 60_dog.prune.in --recode vcf-iid --out 60dog_geno0.05_prune_in "----extract prune in SNP done----"
- 计算样本间IBS距离,这里会生成.mdist.id和.mdist两个文件。.mdist.id文件记录样本名称,.mdist文件记录样本间IBS距离
time $plink --vcf 60dog_geno0.05_prune_in.vcf --distance 1-ibs --double-id --dog --autosome --out 60dog_geno0.05_prune_in - 将.mdist.id和.mdist两个文件处理成MEGA软件可以读入的.meg格式,处理代码如下
#!/usr/bin/python
import sys,re,os
### 读取样本id
ID_file = open(r"60dog_geno0.05_prune_in.mdist.id",""r")
arr = []
for i in ID_file:
arr.append(i.strip().split()[0])
head = map(str,range(1,len(arr)+1))
ID_file.close()
### 读取样本间IBS距离
dist_file =open(r"60dog_geno0.05_prune_in.mdist","r").readlines()
print ("#mega\n!Title: fasta file;\n!Format DataType=Distance DataFormat=LowerLeft NTaxa=%d;\n" % len(arr))
for i,j in enumerate(arr):
print ('['+str(i+1)+']'+' #'+j)
#print
print ('[ '+' '.join(head)+' ]')
print ('[1]')
for l,m in enumerate(infile2):
tmp = m.strip().split()
#tmp[-1] = ''
print ('['+str(l+2)+'] '+' '.join(tmp))
dist_file.close()
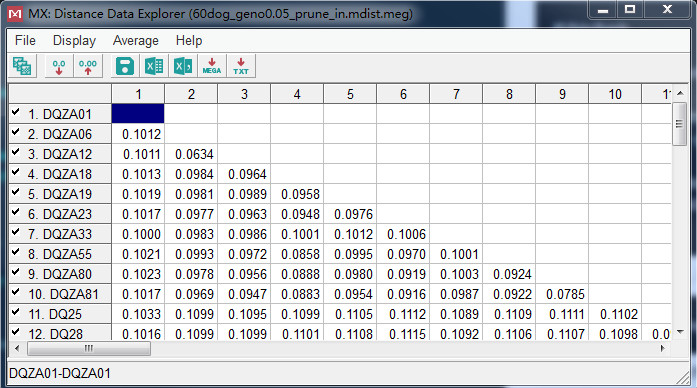
- 将上面生成的.meg文件导入到MEGA软件中,可以看到数据长这个样子
3. 系统发生树可视化
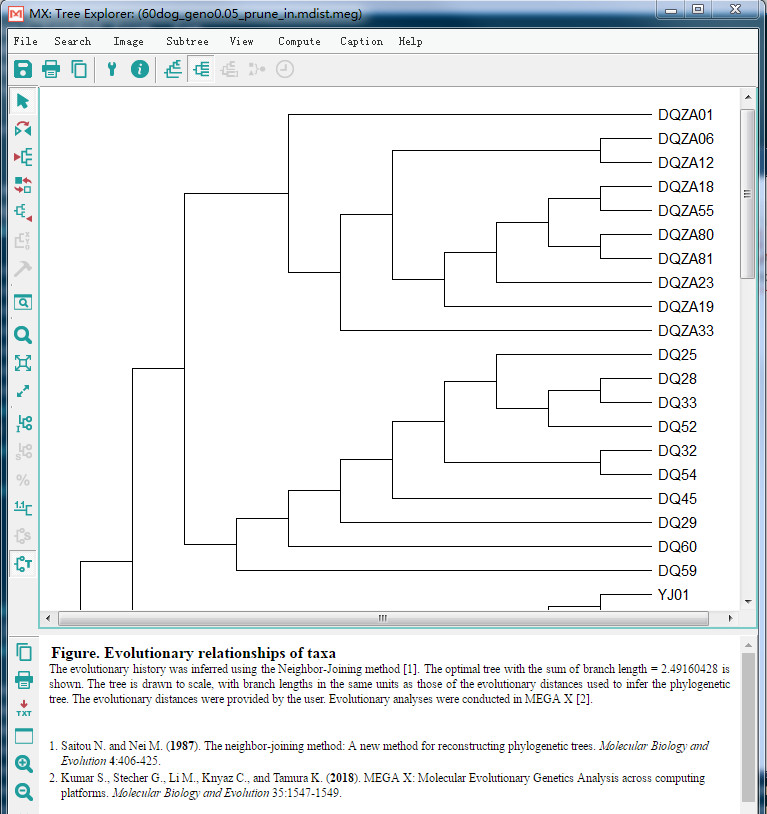
- 在MEGA软件中,读取数据后,选择NJ法进行构建进化树,在弹出的窗口中可以看到树形轮廓基本构建出来
- 接下来将树形图导出为nwk格式文件,进行下一步美化。nwk文件记录样本间亲缘关系相对远近,可以被大部分树形图软件识别。可以看一下我们这里的nwk文件。直接输出在这里,60个样本。
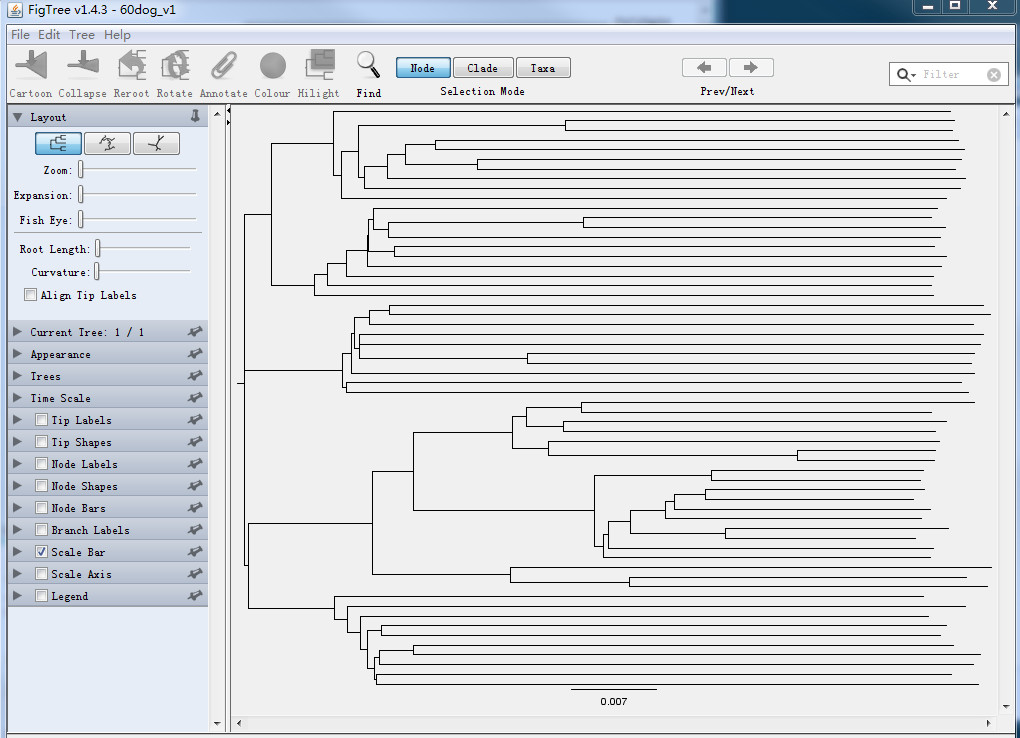
(((DQZA01,(((DQZA06,DQZA12),((((DQZA18,DQZA55),(DQZA80,DQZA81)),DQZA23),DQZA19)),DQZA33)),((((((DQ25,((DQ28,DQ33),DQ52)),(DQ32,DQ54)),DQ45),DQ29),DQ60),DQ59)),(((((YJ01,YJ10),YJ03),((YJ08,YJ16),(YJ14,YJ26))),YJ17),(YJ15,YJ21)),((((((KD106,KD117),(KD110,KD113)),(KD107,(KD207,KD208))),((DKD301,DKD302),((((((DKD303,DKD307),DKD304),DKD306),(DKD305,DKD309)),DKD308),DKD310))),(KD215,(KD222,KD223))),(WM3,(YL1,(LJ6,((YL5,YL7),(((LJ1,LJ2),LJ12),(LJ13,LJ8)))))))); - 使用FigureTree软件进行美化。首先用FigureTree打开nwk文件
然后,选择树形为针叶状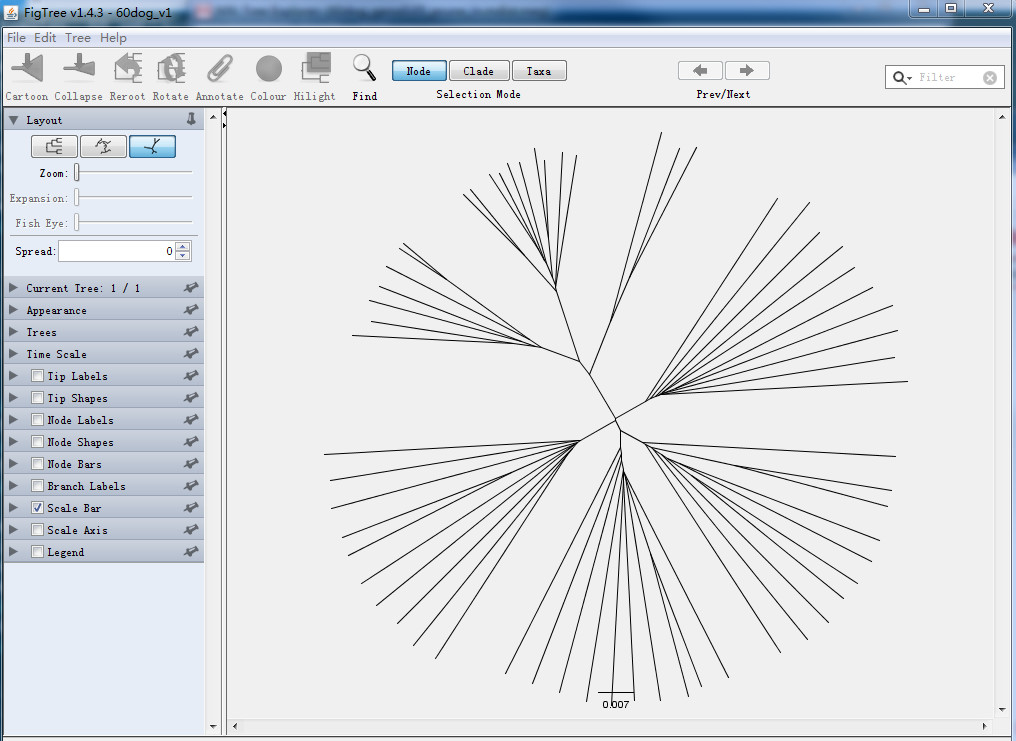
最后为不同子群样本分配颜色,就可以得到文章发表级别的图片了。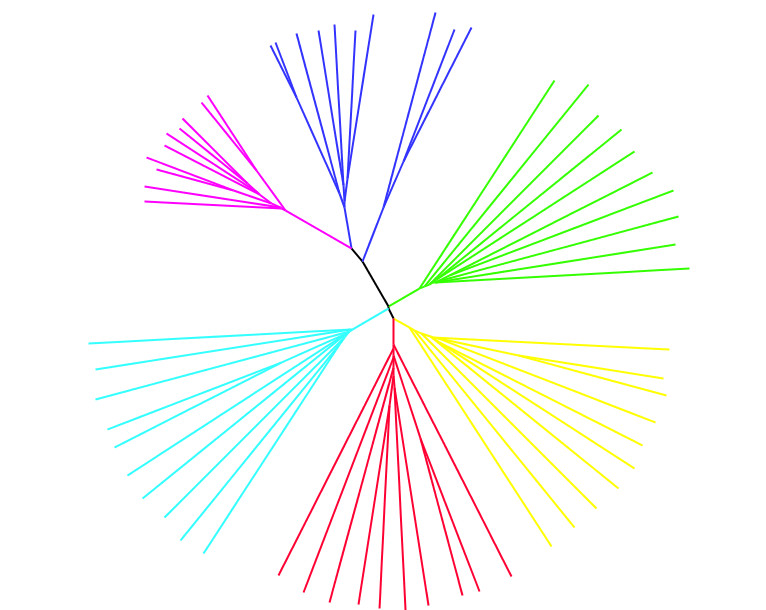
小结
这里构建的是物种树,和基因树有所不同。基于单个同源基因差异构建的系统发生树应称之为基因树,因为这种树代表的仅仅是单个基因的进化历史,而不是它所在物种的进化历史。物种树一般最好是从多个基因数据的分析中得到。
此外,NJ法构建发生树适用于进化距离不大的样本,这也算是缺点之一。
三、 祖先成分分析
祖先成分分析主要是探究个体的混血程度和群体间的基因交流程度。目前,可以做祖先成分分析的软件主要有 STRUCTURE,frqppe 和 admixture。 admixture运算速度快,操作简单,成为了主流的分析软件。
软件下载链接 admixture
1. 简单介绍admixture算法原理
admixture用于从多位点SNP基因型数据集对个体祖先进行最大似然估计,虽然与STRUCTURE使用相同的统计模型,但其快速数值优化算法可以更快速地进行计算估计。具体来说,admixture使用 block relaxation 方法来交替更新等位基因频率和血统比例参数。通过解决大量独立凸优化问题来处理每个block更新,而这些凸优化问题则使用快速序列二次规划算法来解决。通常来说,SNP基因型数据集中的个体应该是不相关的,例如case-control相关性研究中的个体。
2. 数据处理和祖先成分估计
数据过滤
plink=/software/biosoft/software/plink/plink time $plink --vcf 60_dog.merge.vcf --dog --geno 0.05 --maf 0.05 --hwe 0.0001 --make-bed --out 60_dog_qc && echo "----QC and vcf2bed done"估计最优祖先个数,即群体分为几个亚群更合理,这里亚群数目称为K。通常情况下,由于不知道这个群体实际包含几个亚群,可以预设K的范围为2~n。软件就会模拟在K=x的情况下,推算群体是如何分群的,以及每个个个体的血统构成比例。对每个K值模拟的结果,软件都会计算出一个CV error值和最大似然值,error值和最大似然值两个指标都可以挑选最佳K值。对于error值来说
admixture=/software/biosoft/software/admixture_linux-1.3.0/admixture
for K in {2..5}
do
time $admixture --cv 60_dog_qc.bed $K |tee log${K}.out && echo "----admixture K=$K done----"
done
对每个K值模拟的结果,软件都会计算出一个CV error值和最大似然值,error值越小越好,似然值越大越好,两个指标都可以挑选最佳K值。一般而言就是要挑选出error值最小和K值最小的分群策略或者挑选出似然值最大和K值最小的分群策略,即”拐点”。然而,目前学界对于如何判断分群最佳仍有很大争议,CV error值和最大似然值只是参考,无法起决定性作用,因为,目前主流做法是把K值(2~n)都画出来,分别解释。这里取K=2~5,来看下具体结果。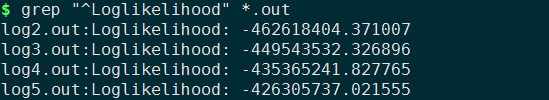
很不幸,这里error值和似然值都是单调的,没有所谓的拐点,可以都画出来看一下。
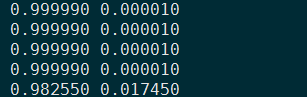
- admixture运行结束后,会产生.Q文件,记录每个个体不同群体祖先成分信息,以K=2为例截图看下
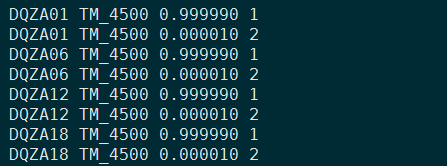
添加上样本ID和所属分组ID,进行可视化。这里以K=2时,来看下处理后的文件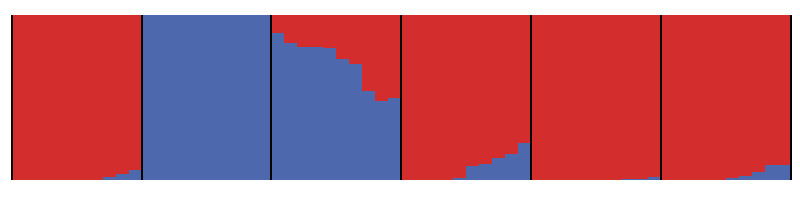
3. 祖先成分堆叠图可视化
这里使用R完成祖先成分堆叠图可视化,由于篇幅限制,这里只放主体部分
library(ggplot2)
library(cowplot)
plotFile<-read.table("dog_qc.2.sample.pop.q.plot",header=f,stringsasfactors = f) colorarr<-c("#d32d2d","#4e68ae") sortplot<-plotfile[order(plotfile[,2],plotfile[,3]),] ##根据样本所属分组和祖先成分比例进行排序 sortplot[,4]<-as.factor(sortplot[,4]) pop<-sortplot[!duplicated(sortplot[,2]),2] ##提取出所有群体id knum<-length(plotfile[!duplicated(plotfile[,4]),4]) ##统计亚群数目 ##统计每个分组起始位置 popindex<-c(0) for(i in pop) { tmp<-which(sortplot[,2]="=i)" popindex<-c(popindex,tmp[length(tmp)]) } popindex<-popindex knum sample<-sortplot[!duplicated(sortplot[,1]),1] ##提取样本id sortplot[,1]<-factor(sortplot[,1],levels="sample)" ### 画堆叠图 p2<-ggplot(sortplot,aes(x="V1,y=V3,fill=V4))+" geom_bar(stat="identity" ,width="1)+" theme(panel.grid.major="element_blank()," panel.grid.minor="element_blank()," panel.background="element_blank()," axis.ticks="element_blank()," axis.text="element_blank()," axis.title="element_blank())+" guides(fill="FALSE)+" scale_fill_manual(values="colorArr)" 画分组标志线 popindex) x="i+0.5" p2<-p2+annotate("segment",x="x,xend" x,y="0,yend" 1,size="1,colour=" black"," arrow="arrow(ends" "both",length="unit(0," cm")))" < code>所以K=2时,堆叠图是这样的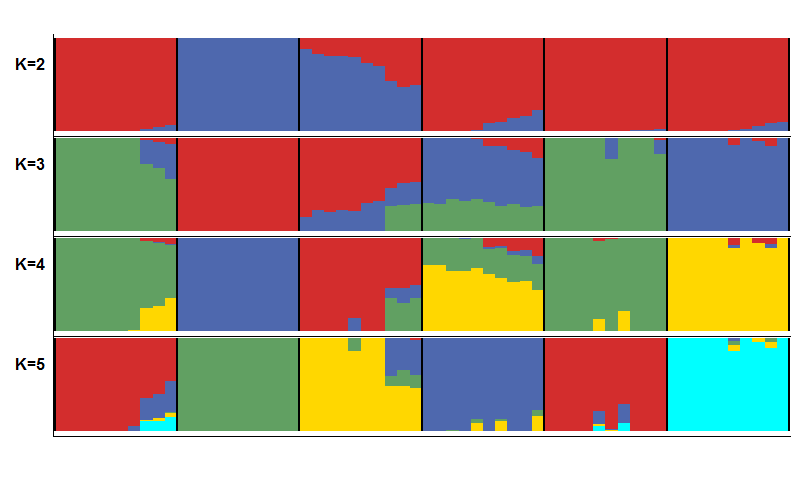
按照上述代码,依次画出k=2~5的堆叠图p2,p3,p4,p5,并使用R包cowplot 组合在一起,代码如下
combine_bar<-ggdraw() + draw_plot(p2,0.05,0.7,0.95,0.25) draw_plot(p3,0.05,0.5,0.95,0.25) draw_plot(p4,0.05,0.3,0.95,0.25) draw_plot(p5,0.05,0.1,0.95,0.25) draw_plot_label(c("k="2" ," "k="3" c(0, 0, 0), c(0.9, 0.7, 0.5,0.3), size="12)" combine_bar < code>合并之后,可视化就基本完成了。
小结
在实际项目中,这三种分析方法得到的结果通常放在一起来看。三种分析均采用不同算法,从不同角度对群体结构进行解释,理解样本间亲缘关系,可以通过PCA散点图和系统发生树来观测样本的分布,但是要了解分组间的基因交流程度和个体混血程度就要依赖祖先成分分析了。三种分析方法相辅相成,可以帮助我们更好的理解样本间的关系,剔除离群样本。



