熊东彦 universebiologygirl
写在前面的话
生物学数据分析中相关性分析是十分重要的一个环节,但这个环节又经常被忽视。以生物信息学分析为例:非生物信息专业的学生通常在分析实验获得测序数据时都会忽略这个问题。
大多数非专业生物信息学背景学生的生物信息数据分析技巧都是从网上的一些教程或者是培训机构的视频中学到的,遗憾的是培训机构仅仅只是教会了你跑一个组学分析的流程,其中涉及的很多重要的细节并没有教给你。因此,从细节着手进行相关性分析显得尤为重要。例如:在转录组测序时候通常要选择3个生物学重复。但是选择了3个生物学重复后送去测序,拿到数据跑完一遍流程真的就可以用来进行差异表达分析吗?绝大多数情况下是可以的,但如果你研究的是细菌、病毒这些变异相对较多的物种,你的一个处理的不同生物学重复可能就不一定是一致的了。如果这种情况发生,那就要剔除这组变异数据。用剩下的数据进行差异表达分析。毕竟设置3个生物学重复一方面是保证一定的重复性,一方面也是预防某个重复值不能用的情况。当然在其他生物学数据分析中,也有这种情况。
今天介绍一个利用R语言编程来进行相关性分析的方法。毕竟当样本数据量较大的情况,手动挨个做相关性分析非常费时间。同时用R语言做完相关性分析后的最大特点是可视化,可以直接提升文章数据的档次感。
手把手教程呈现:
回顾一下相关性分析概念:相关性分析是指对两个或多个具备相关性的变量元素进行分析进而衡量两个变量因素的相关密切程度。相关性的元素之间需要存在一定的联系或者概率才可以进行相关性分析。相关性分析分为pearson相关分析、spearman相关性分析和kendall相关性分析。本次主要介绍Pearson相关分析。它适用于变量为连续性变量且变量之间存在线性关系的情况,二者缺一不可。
示例数据:来源于小鼠感染某种烈性病毒前后的转录组测序数据。实验设计为随机选择3只同一窝出生的小鼠,先提取血液进行转录组测序,随后同时向同窝小鼠注射某种烈性病毒,感染病毒第7天提取血液再进行转录组测序。拿到测序数据后跑完部分转录组分析流程,获取基因表达的reads计数结果。
在进行Pearson相关分析前,按照Pearson相关分析的基本要求先判断两个变量之间是否存在线性关系。随机选取同一个处理的两个生物学重复,用ggplot2绘制两个变量之间的散点图。
library(ggplot2)
ggplot(data = OUT883_withoutgenename[,1:2],mapping = aes(x=OUT883_withoutgenename[,1],y=OUT883_withoutgenename[,2],color="red"))+geom_point()+geom_smooth(method = lm) #geno_point()绘制散点图,geom_smooth()进行拟合,method选择lm是线性拟合
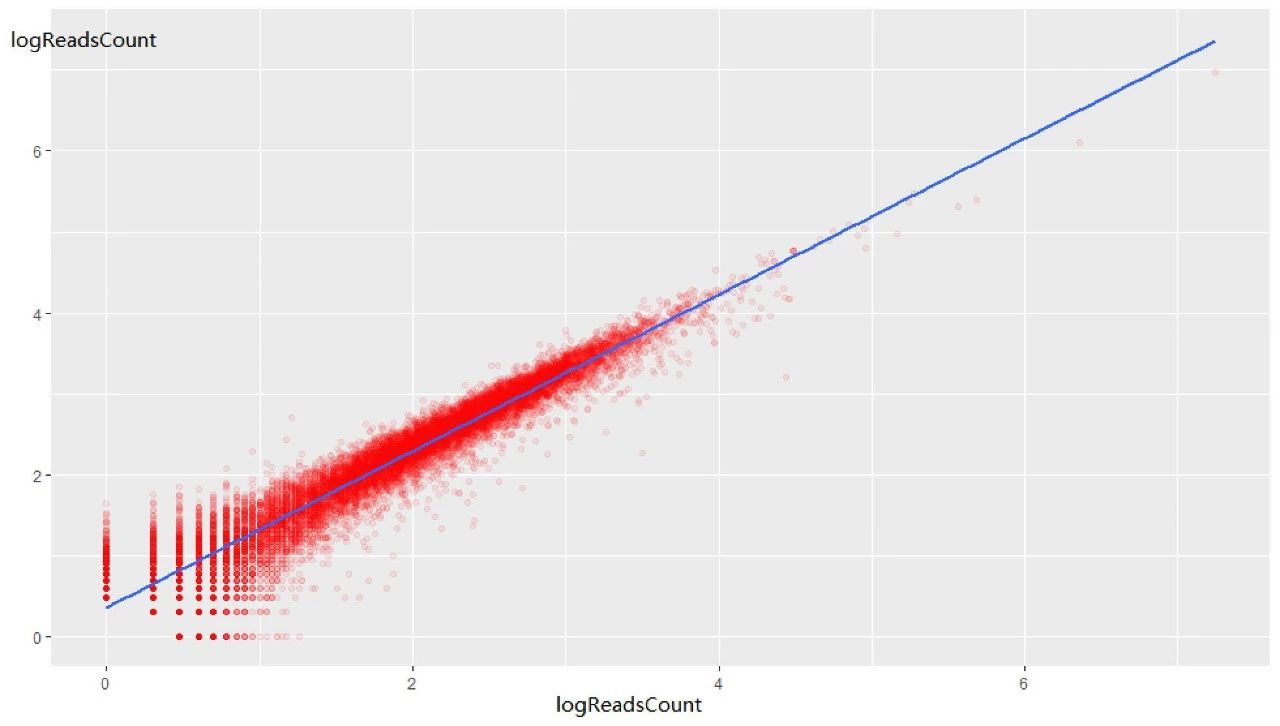
从上图可以看出,除去个别区间,两个变量主要呈现线性关系。之所以个别区间如x=0附近是这样的关系是由于我没有对数据进行过滤。(比如因为测序误差导致的在两个生物学重复之间由于测序仪误差一个基因在被测到而在对应生物学重复内没有测到)。
判断符合Pearson相关性分析条件后可以进行后续分析。现在要对这些数据进行两两相关性分析,以初步判断样本之间是否有较大差异。
readscount<-read.table("病毒感染小鼠前后基因表达统计.txt",header=t,sep="\t")#读取数据 ge_cormatrix<- matrix(ncol="ncol(readscount)-1,nrow" = ncol(readscount)-1)#定义相关系数矩阵 colnames(ge_cormatrix)="colnames(readscount[,2:ncol(readscount)])#定义相关系数矩阵列名" rownames(ge_cormatrix)="colnames(readscount[,2:ncol(readscount)])#定义相关系数矩阵行名" for(i in 1:ncol(ge_cormatrix)){ ge_cormatrix[i,i]<-1#任何一个样本与自身的相关性为1 } readscount_withoutgenename<-readscount[,2:ncol(readscount)]#将读取的数据数值部分赋值给新矩阵 readscount_withoutgenename<-readscount_withoutgenename[rowsums(readscount_withoutgenename)>1,]#过滤掉所有样本的基因表达都为0的行
for(i in 1:(ncol(readscount_withoutgenename)-1)){
for(j in (i+1):ncol(readscount_withoutgenename)){
a<-readscount_withoutgenename[,i] b<-readscount_withoutgenename[,j] t<-cor.test(a,b,method="pearson" ) #使用cor.test函数的pearson模型计算相关系数 cor_num<-as.numeric(t$estimate) #将计算出的相关系数以数值形式传递给cor_num ge_cormatrix[i,j]="cor_num" #将相关性系数赋值给相关系数矩阵对于的位置 ge_cormatrix[j,i]="cor_num" #由于相关系数矩阵是一个对称矩阵,其对称位置也为相同的相关系数 } library(corrplot) corrplot(ge_cormatrix, type="upper" , order="hclust" tl.col="black" tl.srt="45)#使用corrplot函数可视化相关系数矩阵" < code>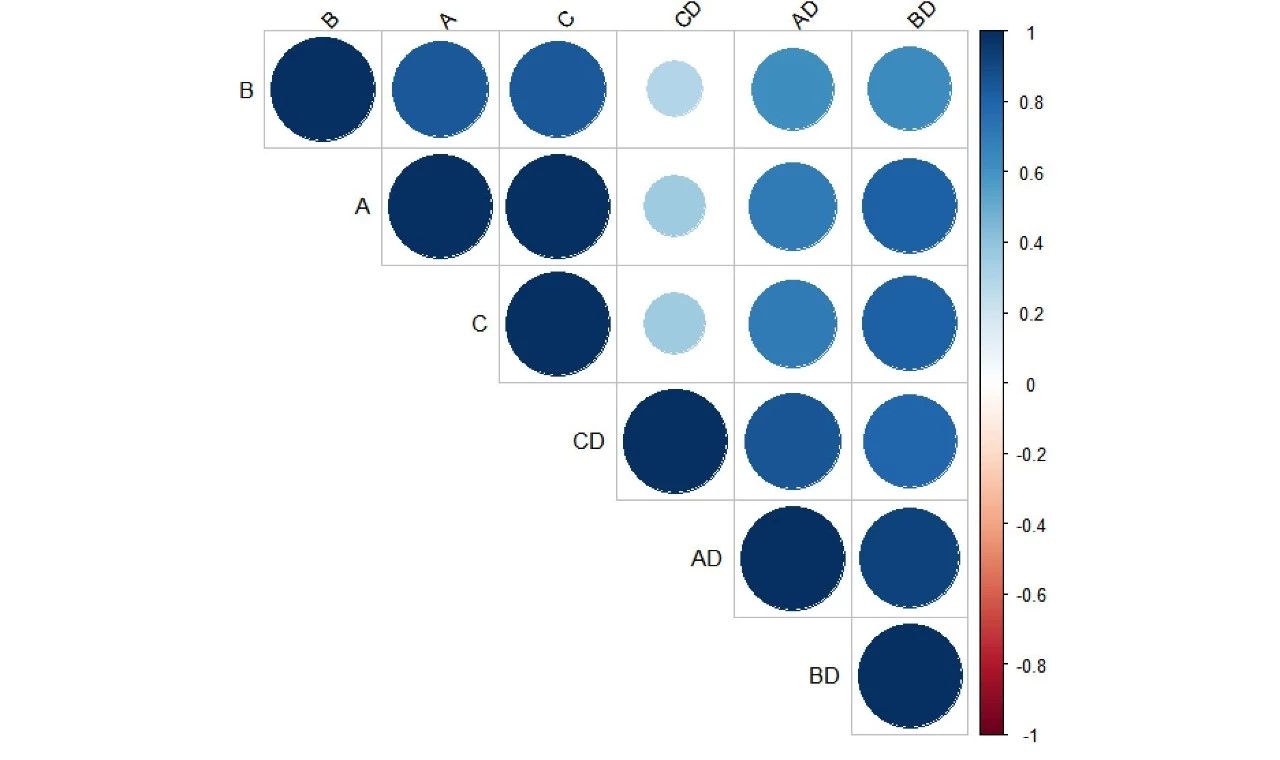
其中ABC指代小鼠感染病毒前的三个生物学重复样本,AD/BD/CD是感染病毒后安乐死的三个生物学重复,从图中结果看出,处理前的三个重复样本相关性高,处理后的三个样本相关性高。可以分别作为重复组进行下游分析。
延伸讨论
本例中有两个可拓展内容,一是本文介绍了三种相关系数计算方法,而作为教学目的只重点介绍了Person相关系数计算方法,另外两种相关系数Spearman和Kendall相关系数用于对分类变量的数据或变量值的分布明显非正态或分布不明时使用,计算时先对离散数据进行排序或对定距变量值排(求)秩。当然最好先通过作图柱状图或者散点图判断一下数据的分布是否属于正态分布,如果属于正态分布而且为连续型变量选择pearson即可,如果不属于就选择另外两个之一。本文中是对转录组基因表达定量的计算,基因表达可以看做正态分布。所以才选择的Pearson相关分析。另外本文的代码部分计算Pearson相关系数使用的函数是cor.test()函数,这个函数通常需要输入三个参数,即两个要计算相关关系的变量(无所谓前后,因为相关性分析只是判断两个变量是否相关),第三个参数是method,可以选择person、spearman、kendall。
Take Home message
用R语言做重复相关性分析重点在于构建相关性矩阵然后利用corrplot函数可视化,要依据自身数据的统计学特点选择合理的相关性模型。



