原创:丸子 宇宙实验媛
由于基因表达调控机制的复杂性,从不同的层面探究生物问题越来越重要,因此需要我们对多种组学数据的整合分析。从RNA-Seq层面,我们可以探究哪些基因具有显著差异,上调或下调;但是想进一步探究调控某一生物学过程的关键因子(包括顺式调控元件和转录因子),以及哪个转录因子调控了感兴趣的基因,需要结合ChIP-Seq来分析。从ChIP-Seq层面,我们可以研究某个特定转录因子的调控作用,以及调控区的组蛋白修饰等。今天,我们就来学习下ChIP-Seq的实验部分,之后的推送也会为您献上ChIP-Seq的数据分析。
概念
染色质免疫共沉淀技术(Chromatin Immunoprecipitation,ChIP)是一种用于研究蛋白质DNA的体内相互作用的经典实验技术。采用特异性抗体将目的蛋白进行免疫沉淀,由此可以把目的蛋白所结合的基因组DNA片段也富集下来。通过与高通量测序技术的结合,对ChIP后的DNA产物进行测序分析,从全基因组范围内寻找目的蛋白的DNA结合位点,以高效率的测序手段得到高通量的数据结果。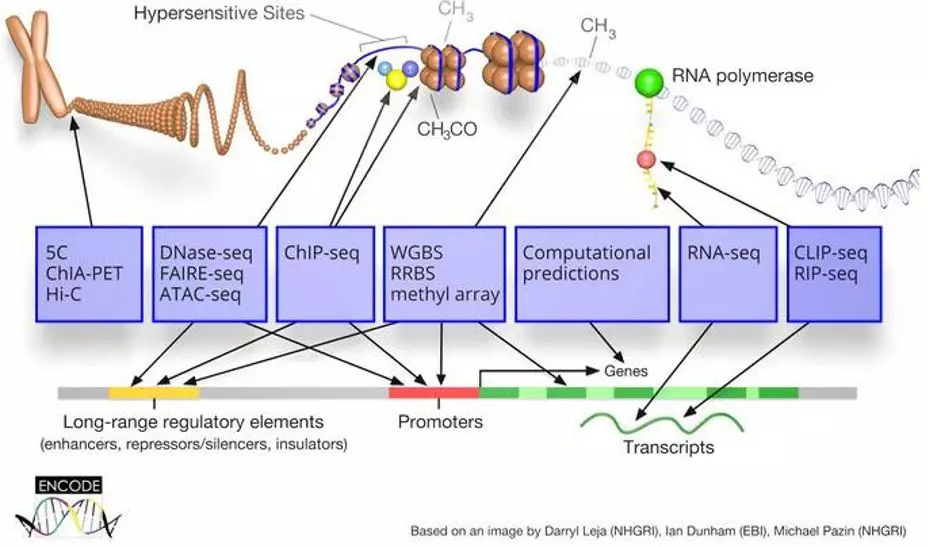
ENCODE数据总览
技术原理
在生理状态下,把细胞内的DNA与蛋白质交联(Crosslink)后裂解细胞,分离染色体,通过超声或酶处理将染色质随机切割; 利用抗原抗体的特异性识别反应,将与目的蛋白相结合的DNA片段沉淀下来; 再通过反交联(Reverse crosslink)释放结合蛋白的DNA片段; 纯化; 测序获得DNA片段的序列,最后将这些DNA片段比对到对应的参考基因组上。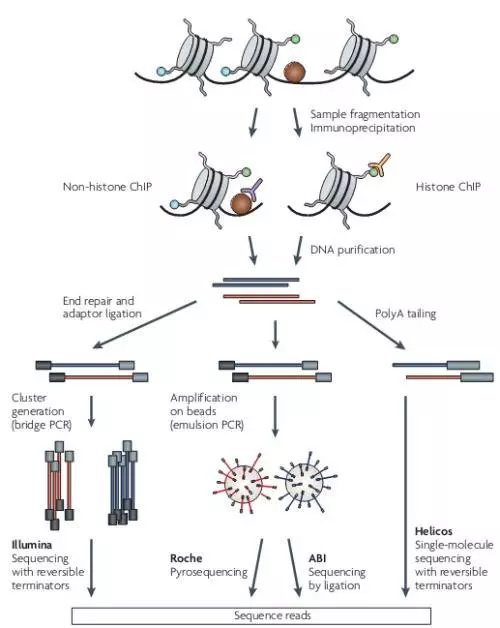
ChIP实验原理
应用领域以及技术优势
由于 ChIP-Seq 的数据是 DNA 测序的结果,为研究者提供了进一步深度挖掘生物信息的资源,研究者可以在以下几方面展开研究:
(1)判断 DNA 链的某一特定位置会出现何种组蛋白修饰;
(2)检测 RNA polymerase II 及其它反式因子在基因组上结合位点的精确定位;
(3)研究组蛋白共价修饰与基因表达的关系;
(4)转录因子研究。ChIP-Seq能够在全基因范围内捕获转录因子或者表观修饰标记结合的目标DNA,鉴定转录因子结合位点,揭示基因调控网络,并且适合多种多样的样本。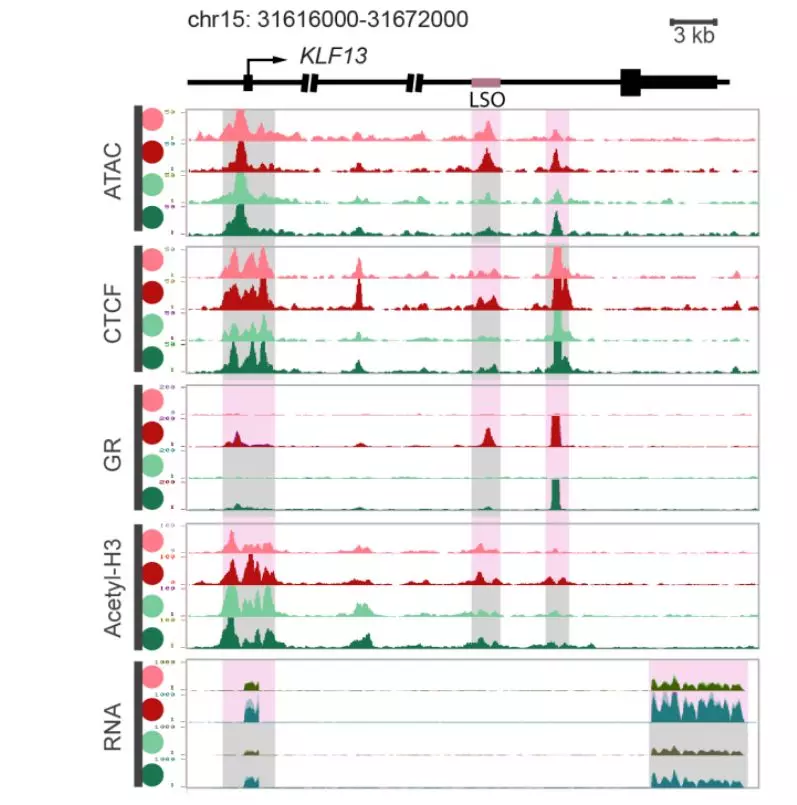
表观遗传修饰与转录调控
实验流程
(1)甲醛处理细胞,使DNA-protein的相互结合作用被交联固定
(2)裂解细胞,得到全细胞的裂解液
(3)超声处理或者用限制性内切酶处理,将基因组DNA打断至100-500bp
(4)抗体免疫沉淀,在细胞裂解液中加入一抗和beads,进行孵育
(5)采用合适的实验条件进行洗脱,并进行解交联
(6)通过qPCR对ChIP结果进行验证
(7)准备好的ChIP后的DNA样品用于ChIP-Seq建库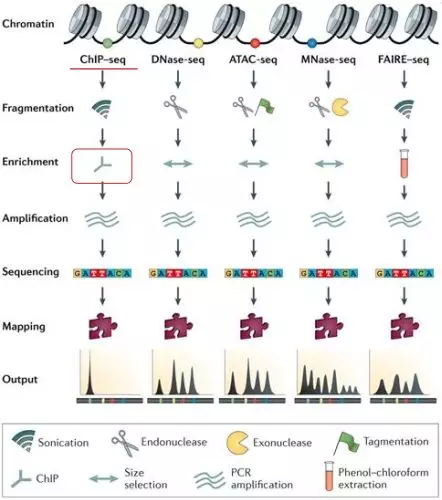
ChIP实验流程
建库流程
(1)DNA片段末端修复
(2)3’端加A碱基
(3)连接测序接头
(4)PCR扩增及DNA产物片段大小选择(一般为100-300bp,包括接头序列在内)
想要了解详细步骤,请参考诺唯赞公司VAHTS™ Universal DNA Library Prep Kit for Illumina® V3。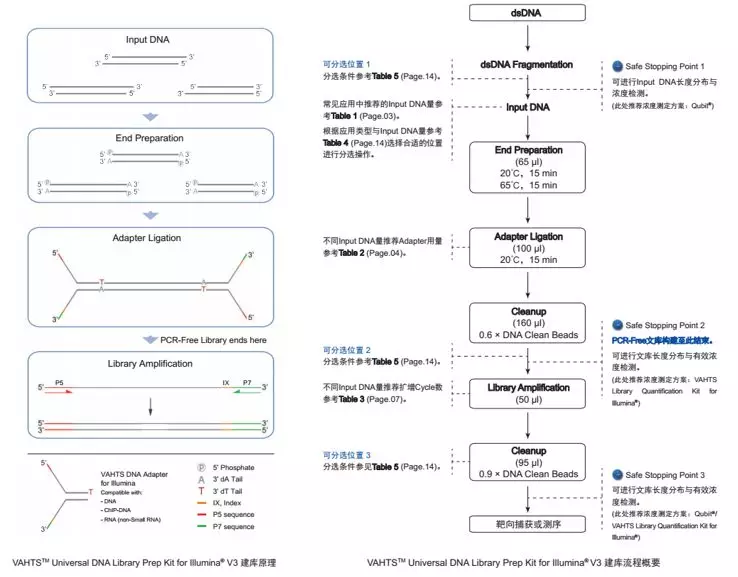
VAHTS™ Universal DNA Library Prep Kit for Illumina® V3建库原理以及流程VAHTS™ Universal DNA Library Prep Kit for Illumina® V3建库 Input DNA量最低可至100 pg,且DNA片段末端修复&加A尾,一步完成。经过了严格的质量控制和功能验证,最大程度上保证了文库构建的稳定性和重复性。
注意事项
- 10^6~10^7 个细胞才能保证最终得到10到100ng ChIP DNA。
一般10^6可以满足高丰度蛋白(如RNA polymerase II)和局部组蛋白修饰(如H3K4me3)的ChIP。如果是低丰度的转录因子蛋白和其他组蛋白修饰则需要10^7个细胞。 - 超声处理在含有SDS的缓冲液中可能会破坏蛋白质-蛋白质和蛋白质-DNA相互作用。
但是含有SDS的缓冲液能增加超声的效率,适应与DNA紧密结合的转录因子的ChIP-Seq,如果结合较弱的话不推荐使用加SDS的缓冲液。 - ChIP 样品中如含有明显的蛋白质或离子浓度过高或其它杂质污染,可能会使库检时 2100峰图异常,对建库过程中的酶反应产生影响,导致建库失败。建议在完成ChIP实验后,选择某一已知的阳性DNA结合区域设计Q-PCR实验,由此验证ChIP实验的可靠性。但此建议不适合于没有阳性对照序列的ChIP实验。
- IgG通常pull down非常少的DNA,这样导致在后期的建库过程中PCR Cycles 数增加,导致不能达到作为control去除背景噪音的目的(会缺失和放大部分信息)。因此比较而言,Input更适合作为control。建议用不同公司的抗体来做生物重复,以避免抗体导致的结果差异,保证结果的准确性。
参考文献
1.Jothi et al. (2008) Genome-wide identification of in vivo protein–DNA binding sites from ChIP-seq data. Nucleic Acids Res 36(16) 5221–5231.
2.Bernstein, BE; et al. (2005). “Genomic maps and comparative analysis of histone modifications in human and mouse”. Cell. 120: 169–181.
3.Johnson, DS; Mortazavi, A; et al. (2007). “Genome-wide mapping of in vivo protein–DNA interactions”. Science. 316: 1497–1502.



